Fuente:
http://www.agenciasinc.es/Noticias/Combaten-la-anemia-aplasica-con-una-terapia-para-retrasar-el-envejecimiento
Investigadores del CNIO han descrito un nuevo tratamiento basado en llevar el gen de la telomerasa a las células de la médula ósea mediante terapia génica, una estrategia para el tratamiento de la anemia aplásica. Los resultados se publican en la revista Blood.
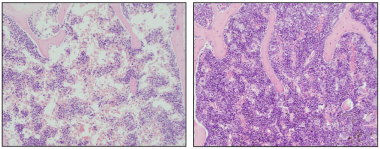
Imágenes representativas de médula ósea con anemia aplásica (izquierda) y médula ósea curada tras el tratamiento con terapia génica con telomerasa (derecha).
La anemia aplásica es una enfermedad rara de la sangre, potencialmente mortal, en la que la médula ósea es incapaz de generar células sanguíneas al ritmo adecuado. Muchas formas de anemia aplásica comparten un importante vínculo con el proceso de envejecimiento: el acortamiento de los telómeros, las estructuras que protegen los extremos de los cromosomas.
Hace cuatro años, un grupo del Centro Nacional de Investigaciones Oncológicas (CNIO) creó una novedosa terapia contra el envejecimiento basada en reparar los telómeros. Ahora, los mismos investigadores demuestran que esa terapia puede ser efectiva contra las clases de anemia aplásica provocadas por telómeros cortos. Es una estrategia del todo nueva en la patología.
El tratamiento se basa en hacer que las células de la médula ósea expresen la enzima telomerasa, responsable de reparar los telómeros. Para ello, los investigadores recurren a la terapia génica: utilizando un virus como ‘taxi’, introducen el gen de la telomerasa en las células de la médula ósea, que pueden así reparar los telómeros y seguir generando células sanguíneas.
“Aportamos una prueba de concepto de que el tratamiento con telomerasa (…) tiene un efecto terapéutico en la anemia aplásica provocada por telómeros cortos”, escriben los autores, entre los que están Christian Bär como primer autor y Juan Manuel Povedano, en una publicación en la revista Blood.
El trabajo ha sido dirigido por Maria A. Blasco en el CNIO y en el que han colaborado la Universidad Autónoma de Barcelona y Hoffmann-LaRoche (Basilea).
La anemia aplásica puede deberse a mutaciones heredadas en —que se sepa por ahora— unos treinta genes, varios de ellos implicados en el mantenimiento de los telómeros. También puede adquirirse por mecanismos aún no claramente establecidos —desde la exposición a determinadas toxinas hasta infecciones víricas—. Un rasgo frecuente en la anemia aplásica, independientemente de si es heredada o adquirida, es la presencia de telómeros cortos.
En 2012 el Grupo de Telómeros y Telomerasa del CNIO, dirigido por Maria A. Blasco, ideó una estrategia para reparar los telómeros. El acortamiento de los telómeros es un proceso que ocurre de forma natural en el organismo cada vez que las células se dividen: durante la división celular, el ADN, empaquetado formando los cromosomas, debe duplicarse, pero el propio diseño de la maquinaria de copia impide replicar completamente el final de los cromosomas. Los telómeros, por tanto, se acortan un poco con cada división celular. Como norma general, a mayor edad, más cortos los telómeros.
La telomerasa, la enzima que repara los telómeros, solo está activa durante el embarazo; es decir, en el organismo adulto las células sanas no expresan telomerasa. Con una excepción: las células madre presentes en los tejidos —en la médula ósea, en el caso de la sangre—. Las células madre, que tienen que dividirse a menudo para regenerar los tejidos con nuevas células, sí pueden producir telomerasa, pero no tanta como para contrarrestar el acortamiento telomérico que se acumula con el envejecimiento: con el tiempo los tejidos tienen menos células de refresco y pierden capacidad regenerativa.
En su trabajo de 2012 los investigadores retrasaron sustancialmente el envejecimiento de los ratones logrando que sus células, durante un periodo de tiempo, de nuevo produjeran telomerasa. En el trabajo que ahora se publica hacen que las células madre de la médula ósea produzcan más telomerasa, y puedan así reparar sus telómeros excesivamente cortos.
Los investigadores recurrieron a dos tipos de ratones que simulan la enfermedad en humanos. En uno de ellos se reproduce la anemia aplásica adquirida: debido a daños de diversa etiología una parte de las células madre muere, y las que quedan deben dividirse más para mantener la producción de las células de la sangre; a consecuencia de tantas divisiones, los telómeros se acortan y aparece la enfermedad.
En el animal modelo, los investigadores matan parte de las células madre eliminando en ellas un determinado gen; al cabo de unas semanas tratan a los animales con la terapia génica con telomerasa.
“Efectivamente, el tratamiento [con telomerasa] evita de forma significativa la mortalidad por anemia aplásica, y alarga los telómeros en la sangre y en la médula ósea”, comentan los autores. “En el grupo no tratado con terapia génica la mayor parte de los animales mueren de anemia aplásica, y mueren mucho antes”.
El segundo modelo animal busca reproducir la anemia aplásica hereditaria, producida por mutaciones relacionadas con los telómeros y la telomerasa. Los investigadores recurren a un ratón al que se ha eliminado el gen de la telomerasa específicamente en las células de la médula ósea.
Como en el primer caso, tras ser tratado con terapia génica con telomerasa en este ratón “también se alargaron los telómeros en sangre periférica y el número de células sanguíneas mejoró considerablemente”, escriben los autores. “En ambos modelos, el mayor número de células sanguíneas puede atribuirse a una mayor reserva de células madre”.
Hay clases de anemia aplásica que no están asociadas a telómeros cortos. Pero los investigadores consideran que estos resultados son una prueba de concepto de que la terapia génica es una estrategia válida contra anemia aplásica. Podría aplicarse con otros genes —en vez de con telomerasa—, si se descubriera que tienen un papel causal en esas otras formas de la enfermedad.
Referencia bibliográfica:
Christian Bär, Juan Manuel Povedano, Rosa Serrano, Carlos Benitez-Buelga, Miriam Popkes, Ivan Formentini, Maria Bobadilla, Fatima Bosch, Maria A. Blasco. Telomerase gene therapy rescues telomere length, bone marrow aplasia and survival in mice with aplastic anemia. Blood (2016). doi: 10.1182/blood-2015-08-667485