Hasta el 90% de las muertes por cáncer se deben a las metástasis, pero no hay tratamientos contra ellas. Ahora, un estudio ha identificado las células que las inician en varios tipos de tumores. Para su sorpresa, los investigadores han visto que las implicadas dependen de grasas como los aceites de palma o de coco, presentes en las comidas procesadas.
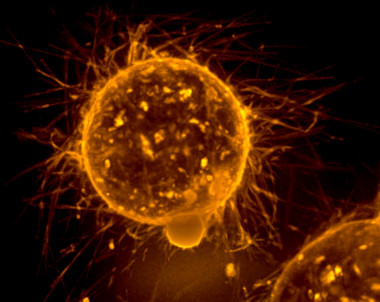
Microtentáculos en la superficie de un tumor de mama.
El cáncer es un enemigo a distancia. Nace y crece en un lugar concreto, pero hasta el 90% de las muertes que provoca dependen de los viajes que sus células emprenden por otros rincones del organismo: las metástasis.
Pero las claves de esas metástasis siguen jugando al escondite en los laboratorios. Aunque había sospechas, ni siquiera se conocían qué células concretas del tumor las iniciaban. Y, lo más importante, no existe ningún tratamiento específico ni eficaz contra ellas.
Ahora, una investigación liderada por el grupo de Salvador Aznar, investigador ICREA en el Instituto de Investigación Biomédica (IRB Barcelona), apunta a una pequeña gran revolución. El estudio que publica su equipo en la revista Nature ha dado tres saltos: identifica las células de origen de la metástasis en varios tipos de tumores; revela que se diferencian del resto porque dependen de las grasas –entre ellas, del ácido palmítico de muchas comidas procesadas–; y da con un marcador que, al inutilizarlo en ratones, previene por completo la formación de metástasis y reduce las ya existentes.
Este ha sido su viaje y hasta dónde han llegado.
“Nosotros no pretendíamos estudiar las metástasis”, comenta Salvador Aznar. “Yo llevaba tiempo analizando las células madre del cáncer y quería profundizar en un tipo particular, las células quiescentes o dormidas”.
Una teoría cada vez más aceptada sostiene que un tumor, igual que un tejido normal, tiene unas células madre que lo originan y a partir de las cuales se derivan todas las demás. Pero dentro de ellas hay un subgrupo compuesto por células madre dormidas, que parecen funcionar como una reserva. "Podrían ser más resistentes a los tratamientos y estar detrás de las recaídas tras la quimioterapia”, añade Aznar.
La sorpresa llegó cuando trataron de separarlas y aislarlas. Usando muestras de pacientes con cáncer oral vieron que había, en efecto, células madre de ciclo más lento. “Pero cuando las analizamos, ellas mismas nos dijeron que tenían algo que ver con las metástasis, porque expresaban muchos genes relacionados con éstas. Parecían adipocitos, células de la grasa. En ese momento cambiamos completamente el objetivo de la investigación”.
Para poder jugar con las células se necesita encontrar una cerradura que las diferencie. De entre todos los genes especialmente activos de las células dormidas había uno que llamaba la atención. Se llama CD36 y la proteína a la que da lugar es una puerta de entrada a las células.
Jugando con ella en ratones, los resultados fueron impactantes: cuando se aumentaba en los tumores orales, que suelen dar metástasis a los ganglios linfáticos en un 20% de las ocasiones, el porcentaje aumentaba hasta un 80% y los ganglios eran 40 veces más grandes. Al contrario, cuando se usaban anticuerpos que la bloqueaban, las metástasis disminuían entre un 80% y un 90%. En algunos casos incluso desaparecían. Y si se administraban antes de introducir las células cancerígenas, prevenían por completo su aparición. Todo ello, curiosamente, sin afectar apenas al tumor de origen.
Al revisar datos de estudios previos, el grupo de Aznar observó que el aumento de CD36 en pacientes con cáncer de pulmón, mama o vejiga también estaba relacionado con un peor pronóstico. Y cuando probaron en los ratones con melanoma o cáncer de mama, los resultados fueron muy parecidos. En el artículo hablan de un mecanismo general de metástasis.

Aznar y Pascual en su laboratorio del IRB.
¿Por qué nadie lo había estudiado antes? “No lo sé”, reconoce Aznar. “Los datos estaban ahí y algunos ya habían mostrado la relación con el pronóstico, pero quizá la gente prestaba atención a aquello que le interesaba. Se parece a lo que ha sucedido con CRISPR, cuya existencia descubrió un español hace años pero hasta mucho después no hubo quien vio las posibilidades y desarrolló la técnica”.
Ahora están trabajando con MRC Technology, del Reino Unido, para desarrollar anticuerpos que puedan probarse en humanos. Ese es el verdadero salto. “Esperamos conseguirlos –comenta Aznar– y que no se queden en el camino. En cualquier caso habrá que esperar unos años”. Un aspecto positivo es que, al menos en ratones y administrados durante periodos no demasiado largos de tiempo, los efectos secundarios no parecen graves. Otro: los ratones no parecían generar resistencias, presentes en la inmensa mayoría de las terapias dirigidas contra el cáncer.
Para Joan Massagué -asesor científico del propio IRB y director del Instituto Sloan Kettering-, que no ha participado en el estudio, “este trabajo es una excelente contribución al creciente conocimiento sobre las células que originan las metástasis”. Entre sus limitaciones, apunta que “está basado casi exclusivamente en metástasis a nódulos linfáticos, que no son las más temibles, y ha sido realizado en ratones desprovistos de inmunidad, que es una barrera fundamental contra ellas”.
Aznar, sin embargo, señala que “también hicimos experimentos donde se veía la relación con metástasis a pulmón, hígado o hueso”. Y aunque la mayor parte de los datos provienen de ratones sin inmunidad, “hicimos algunos ensayos con ratones inmunocompetentes y los resultados eran similares”.
Para Héctor Peinado, jefe del grupo de Microentorno y Metástasis en el Centro Nacional de Investigaciones Oncológicas (CNIO), y que tampoco ha participado en el estudio, “habría que estudiar en detalle cada tipo tumoral para saber si se trata de un mecanismo universal, pero es sin duda un nuevo hit en el campo de la lucha contra la metástasis. Existe información muy limitada sobre las células que las inician. Es un gran paso el hecho de tener un marcador que puede servir de diana terapéutica, conocer el mecanismo implicado y la existencia de una terapia que podría combinarse con las actuales”.
La investigación tiene, por tanto, un doble valor. Por una parte permite identificar las células que inician la metástasis en al menos varios tipos de cáncer, lo que acelerará y mejorará la investigaciones. Por otra, abre la puerta a posibles nuevos tratamientos. Pero hay una tercera parte: la relación con las grasas y nuestro estilo de vida.
El vínculo entre una dieta rica en grasas y algunos tipos de cáncer, como los de colon y mama, es ya conocido, pero no tanto su correspondencia con las metástasis. “Hasta el momento se han descrito algunos estudios que describen la obesidad como un factor de riesgo de metástasis en cáncer de páncreas y algunos tipos de mama, pero los mecanismos implicados se desconocen”, afirma Peinado.

Cuando las células tumorales se incubaban con ácido palmítico, abundante en las carnes, grasas lácteas y aceites de coco y de palma, estas parecían recordar la relación y producir más metástasis tras inocularse en los ratones.
Teniendo en cuenta que las células iniciadoras parecían grasas, y la importancia de CD36 como puerta de entrada, el grupo de Aznar diseñó una serie de experimentos para comprobar cómo podía afectar la dieta de los ratones al desarrollo de metástasis. Los resultados fueron contundentes: había más y mayores metástasis cuando su dieta contenía más grasas. Y no hacía falta llevarla a valores desproporcionados. “Eran el equivalente a lo que llamamos una dieta de cafetería en humanos”, apunta Aznar.
El mecanismo exacto aún se desconoce. “Podría ser que permitan a las células obtener más energía y resistir mejor el estrés que les debe suponer salir de su entorno y colonizar otros tejidos”, sostiene Aznar. Pero “también podrían jugar un papel otros procesos, no solo la obtención de energía”. De hecho, no todas las grasas parecen perjudiciales: por ejemplo, el consumo de aceite de oliva en la dieta mediterránea se asocia con cierta protección contra el cáncer.
De momento hay al menos un sospechoso claro. Cuando las células tumorales se incubaban con ácido palmítico, estas parecían recordar la relación y producir más metástasis tras inocularse en los ratones. El dato es importante: el ácido palmítico está presente en el aceite de palma o de coco, incluidos en muchos de los productos industriales procesados. “No lo sabemos aún, pero este hecho podría estar detrás del aumento de mortalidad en algunos tipos de cáncer que se ha observado en los últimos años”, sostiene Aznar.
Averiguarlo con exactitud es muy complicado. Implicaría diseñar grandes estudios epidemiológicos de muy difícil seguimiento. Una forma indirecta pero más plausible es la que va a iniciar el grupo del propio Aznar. “Tenemos acceso a muchas muestras de pacientes con cáncer. En colaboración con el Hospital Vall d´Hebron, que también participó en este estudio, queremos analizar si hay una relación entre las grasas presentes en sangre y el riesgo de desarrollar metástasis”.
Otra puerta lógica que se abre es la posibilidad de ofrecer tratamientos dietéticos. ¿Podría una dieta baja en grasas mejorar el pronóstico de los enfermos? “Estamos estudiando también hacer un ensayo clínico para averiguarlo”, comenta Aznar. “Pero es complicado. La labor de seguimiento es muy cara y obtener financiación cuando no hay detrás un posible beneficio económico es más difícil”.
De momento el viaje tiene tres partes. El tiempo dirá si llega la cuarta, la que lo confirme.
Referencia bibliográfica:








